
Today, a quick update on my progress with the RDKit converter! It’s already working (with very minor tweaks) for MOL2 files, and any other format that contains bond orders or bond types.
How it works
RDKit requires 2 things to create a complete molecule from scratch:
- Elements (either the symbol of the atom, or the atomic number). This will allow us to create atoms which will also store all the extra information regarding residues, atom names and types, temperature factors…etc.
- Bonds, or more specifically the indices of bonded atoms and the bond type.
Once we have our elements, we start by creating the corresponding atoms with the Chem.Atom class.
We add to the atom all the information that is typically available in PDB files through the Chem.AtomPDBResidueInfo class (atom name, residue name and number, temperature factor…etc.). Other properties (charges, atom types…etc.) are directly stored as an atom property through the atom.SetProp method.
Finally, we create a dictionary mapping MDAnalysis atom indices to the corresponding indices in the RDKit molecule, which we will need when we create bonds.
Here is a simplified example of what happens under the hood:
# Universe
u = mda.Universe(PDB)
mol = Chem.RWMol()
atom_mapper = {}
for atom in u.atoms:
# create atom
rdatom = Chem.Atom(atom.element)
# add PDB-like properties
mi = Chem.AtomPDBResidueInfo()
mi.SetResidueName(atom.resname)
rdatom.SetMonomerInfo(mi)
# other properties
rdatom.SetProp("_MDAnalysis_type", atom.type)
# add atom to the molecule
index = mol.AddAtom(rdatom)
# map index in universe to index in mol
atom_mapper[atom.ix] = index
Of course the real code is a bit more complex to cover all available topology attributes as well as edge cases.
Then we can add the bonds. If they are not present in the MDAnalysis topology, they are automatically guessed using the same algorithm that VMD uses. We also have to convert whatever bond type or bond order is present in the MDA topology to an RDKit Chem.BondType.
RDBONDORDER = {
1: Chem.BondType.SINGLE,
2: Chem.BondType.DOUBLE,
3: Chem.BondType.TRIPLE,
}
for bond in u.atoms.bonds:
bond_indices = [atom_mapper[i] for i in bond.indices]
bond_type = RDBONDORDER.get(bond.order, default=Chem.BondType.SINGLE)
mol.AddBond(*bond_indices, bond_type)
Again, this is a simplified version of what happens in the actual code.
Finally, we just add a Chem.SanitizeMol(mol) and voilà, we have a proper RDKit molecule!
Here is a working example with mol2 files. Since MOL2 files don’t have elements but only atom names and types, we will have to guess the elements first:
>>> import MDAnalysis as mda
>>> u = mda.Universe(mol2_molecule)
>>> # guess missing info
>>> elements = mda.topology.guessers.guess_types(u.atoms.names)
>>> u.add_TopologyAttr('elements', elements)
>>> # convert
>>> mol = u.atoms.convert_to("RDKIT")
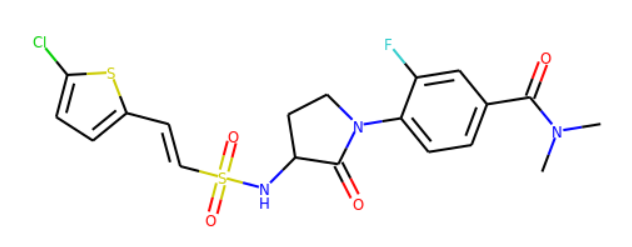
>>> Chem.MolToSmiles(Chem.RemoveHs(mol))
'CN(C)C(=O)c1ccc(N2CCC(NS(=O)(=O)C=Cc3ccc(Cl)s3)C2=O)c(F)c1'
>>> mol
What’s next ?
Most MD topology files don’t explicitely keep bond orders or bond types, but we need them if we want to do anything more complex than an alkane. Nonetheless, bond types and formal charges can be inferred from the connectivity as long as the topology explicitely contains all the hydrogen atoms. Fortunately, as the name states it, “all-atom” simulations have all their hydrogen atoms explicitely written in the topology so we will be able to build the complete molecule with proper charges and bond types from that. This will be the next step once the simple RDKit converter is ready and accepted for merging.
Additionally, the current version doesn’t store any coordinates yet, this will be the last step once everything regarding the topology is done.
Once the coordinates are added, we will be able to retrieve the stereochemistry information for atoms and bonds (R/S, Z/E) base on the 3D structure.
We’re also currently discussing the future API for converters, i.e. do we keep the current u.atoms.convert_to("RDKIT") or do we add something more user-friendly ? Most people seem to be in favor of u.atoms.to_rdkit() for tab-completion, but nothing is decided yet.
You can follow the advancements of this project in PR#2775, and you can already play with the RDKit parser in the develop branch of MDAnalysis.
Cédric