
Last time, I presented a very simple MDAnalysis to RDKit converter which had quite limited applications. Today, I’m proud to announce that the converter can output an RDKit molecule with bond orders and charges from any type of input accepted by MDAnalysis!
Showcase
The only requirement for this to work is having explicit hydrogens in the topology file, which should be the case for most MD simulations.
Let’s start with a simple example: a PDB file.
>>> import MDAnalysis as mda
>>> import nglview as nv
>>> from nglview.datafiles import PDB
>>> u = mda.Universe(PDB)
>>> elements = mda.topology.guessers.guess_types(u.atoms.names)
>>> u.add_TopologyAttr('elements', elements)
>>> u
<Universe with 5547 atoms>
>>> lig = u.select_atoms("resname LRT")
>>> view = nv.show_mdanalysis(lig)
>>> view
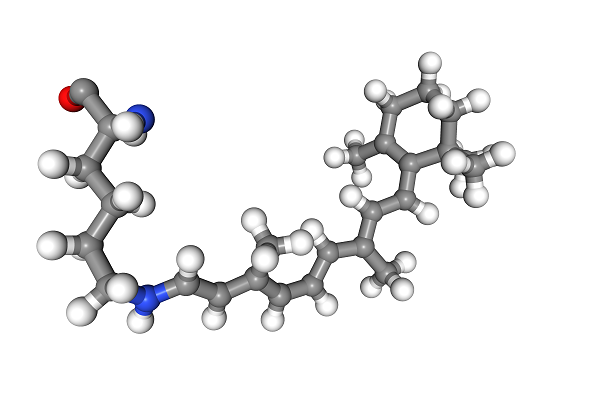
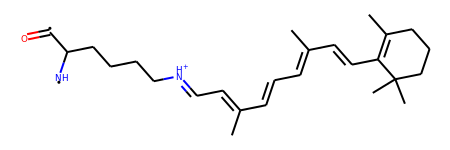
This molecule looks like a derivative of retinal, the compound that binds to opsins and is at the molecular basis of our sense of sight. Now we simply convert the Universe to RDkit and TADA!
>>> mol = lig.convert_to("RDKIT")
>>> Chem.RemoveHs(mol)
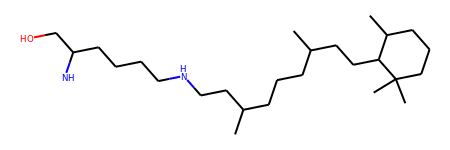
This looks cool, but why would you go through all this when RDKit already reads PDB files ?
The reason is simple: since PDB files don’t contain bond orders most of the time, RDKit will infer them only for standard residue names, not for ligands:
>>> mol = Chem.MolFromPDBFile(PDB)
>>> mol = Chem.SplitMolByPDBResidues(mol)["LRT"]
>>> mol.RemoveAllConformers()
>>> mol
Another example with a topology file from Amber:
>>> from MDAnalysisTests.datafiles import PRM
>>> u = mda.Universe(PRM)
>>> elements = mda.topology.guessers.guess_types(u.atoms.names)
>>> u.add_TopologyAttr('elements', elements)
>>> u
<Universe with 252 atoms>
>>> mol = u.atoms.convert_to("RDKIT")
>>> Draw.MolToImage(Chem.RemoveHs(mol), size=(700, 500))
How it works
Since we have a molecule with explicit hydrogens, we can guess the bond orders and charges from this information. For each atom, we compare it’s expected valence with the current valence to get the number of unpaired electrons. Then their are 3 cases:
- The atom has no unpaired electrons → no need to do anything
- The atom has a too many paired electrons → it needs a positive charge
- The atom has
Nunpaired electrons. In this case, we look at the number of unpaired electrons for each of the neighbors of this atom. If both the atom and his neighbor have unpaired electrons, we increase the order of the bond between them by the smallestN. Finally, if the atom still has unpaired electrons after that, we convert this number to a negative formal charge.
Here’s the process in action:
Now the problem with this method is that the order in which atoms are read might influence the process and incorrectly charge some atoms. Atoms with several possible valences might also get in the way.
For example, a sulfate (SO42-) in a topology would appear as a sulfur atom with 4 connections to the oxygen atoms. Since a valence of 4 is valid for the S atom, we would end up with a sulfur sharing single bonds with 4 negatively charged oxygen atoms.
The way I tackled this problem was to use reactions based on SMARTS patterns to reassign the correct bond orders and charges to atoms.
Here is an example with an arginine, which had a negatively charged carbon instead of a positively charged nitrogen. The corresponding SMARTS reaction is [N;H1:1]-[C-;X3;H0:2](-[N;H2:3])-[N;H2:4]>>[N:1]-[C;+0:2](-[N:3])=[N;+1:4]
Another problem which took me some time to solve was with conjugated systems. For the same reason as above (atom order), in some cases I ended up with negative charges on the atoms at the edges of a conjugated system, and one double bond less than expected.
Now this could also be solved by reactions, but it would mean writing a SMARTS pattern for every possible length of conjugated system: one for 2 alternating double bonds, one for 3…etc.
The good thing is that all the badly written conjugated systems share the same pattern: an anion connected by a single bond to an uncharged atom connected by a double bond to another uncharged atom, a.k.a. [*-]-[*;+0]=[*;+0;!O] as a SMARTS query, then N alternating single and double bonds, and the same 3-atom pattern the other way around.
The idea here was to iteratively decrease N by switching the charges and bond orders of atoms that matched the above 3-atom pattern, a bit like moving up a ladder.
Then at some point we need an end condition to our iterative procedure. It’s triggered when we reach the simplest case where its just 2 anions separated by 2 atoms with a double bond in the middle, which can be solved by a SMARTS reaction.
With the example of an histidine, here’s how the simple case works:
Sometimes unfortunately one of the atoms at the edge of the conjugated system is a nitrogen, which will not be negatively charged since a valence of 3 is expected. We just have to slightly correct the SMARTS query for the end condition.
To detect automatically this rare scenario, we can simply count the number of unique anions that matched the 3-atom pattern [*-]-[*;+0]=[*;+0;!O] and change the SMARTS query when it’s odd instead of even.
Taking our first retinal-like molecule, you will see the carbanion moving up the “ladder” of bonds until it reaches the nitrogen.
What’s next
This bond order guessing took quite some time to implement and test, but at least I’m really satisfied with it now. It overlapped a bit with the time I wanted to spend adding coordinates to the RDKit molecule but this shouldn’t take too long.
Once this is done (probably just before August), the second part of my Google Summer of Code project will finally be over and I can start experimenting with RDKit inside MDAnalysis to import cool features and new kinds of analysis.
It’s gonna be awesome! ![]()
Until then,
Cédric